
Inferring gene regulatory networks (GRNs) from data such as knockout screens or single-cell assays has many applications. In particular, it helps identify biological mechanisms and enhances the discovery power of GWAS studies. New computational methods continue to improve these results.
A gene regulatory network is a directed graph where each arc A → B indicates that gene A has a regulatory impact on gene B. For example, knocking out gene A might increase or decrease the expression of gene B. These interactions can be simplified as chains, e.g., A → C → B additionally represents A → B. Simplifying a large collection of interactions, say E, into a more streamlined network, say F, with fewer arcs can increase our confidence in F‘s interactions, especially when E’s interactions are identified with a potential for false discoveries. This also provides good candidates for direct interactions.
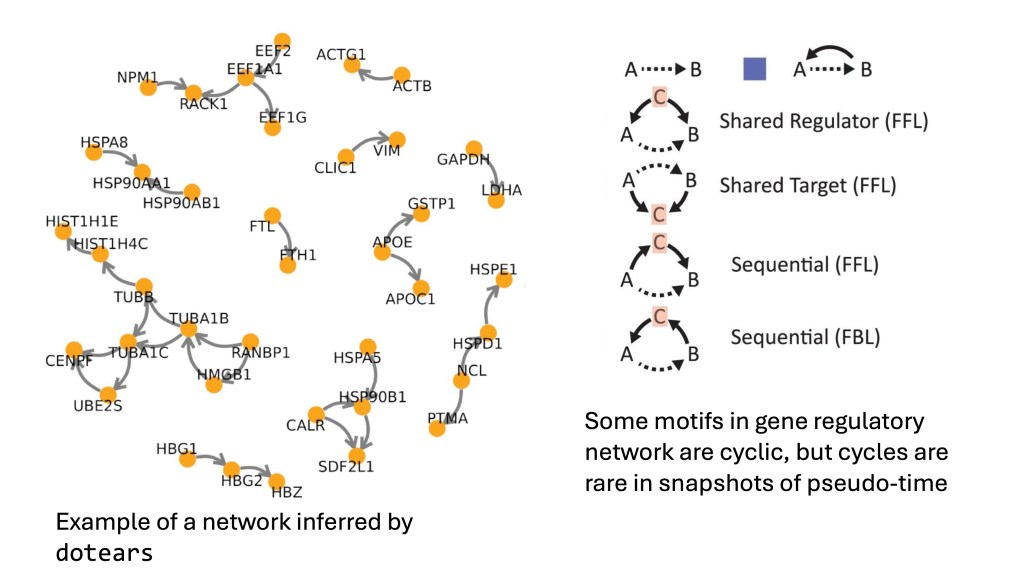
‘Dotears’, developed by Xue et al. and presented at this year’s RECOMB conference, finds acyclic GRNs from interactions inferred from massively parallel CRISPR interventions (individual or multiple gene knockouts). They approach the problem using a continuous optimization technique published in 2018 to represent acyclic networks. According to their reports, in each of four CRISPR studies, they identified over 2000 interactions, 65% of which were validated —- several times more than previous methods.
However, gene interaction networks typically contain cycles, such as feedback loops. While disallowing cycles may statistically enhance predicted interactions, biologically important cycles are lost. Reagor et al. address network discovery using single-cell expression and ATAC assays by mapping cells into ‘pseudo-time’. This approach, related to processes like cell response to stimuli and cell cycle progress, simplifies network identification by naturally excluding cycles. It’s worth noting that this method isn’t applicable to CRISPR interventions, where no natural time progression exists.
References
- Reagor CC, Velez-Angel N, Hudspeth AJ. Depicting pseudotime-lagged causality across single-cell trajectories for accurate gene-regulatory inference. PNAS Nexus. 2023 Mar 30;2(4)
- Xue A., Rao J., Sankararaman S., Pimentel H., dotears: Scalable, consistent DAG estimation using observational and interventional data, arXiv.org/pdf/2305.19215
