
Standard methods sometimes produce nonsense. In 2020, researchers of UC La Jolla published in Nature their discovery that microbiome present in cancer samples identifies their cancer identity and type, using authoritative TCGA data base, hg19 genome assembly and Kraken pipeline to identify microbial reads in TCGA samples.
Currently, a group of (mostly) Johns Hopkins researchers showed that (a) 99.9% of reads attributed to microbes were actually human (b) classifier that was used amplified batch effects, like converting zero counts to differential values between sample types.
The selection of human reference by La Jolla group was totally standard at the time, much more complete genome reference, T2T-CHM13, was published in 2022, using new technology that produces reads longer than 25kbp with high fidelity. However, even before it was widely known that previous releases were incomplete because of technological limitations.
The selection of microbial reference was “standard”, but more problematic: in 2019, alerts started to appear about human DNA included in bacterial genomes. Nevertheless, La Jolla group overlooked that, perhaps they selected references before the alerts and were too busy afterwards, and used the contaminated references from GeneBank to identify types of microbes from reads, using reads not mapped with incomplete hg19.
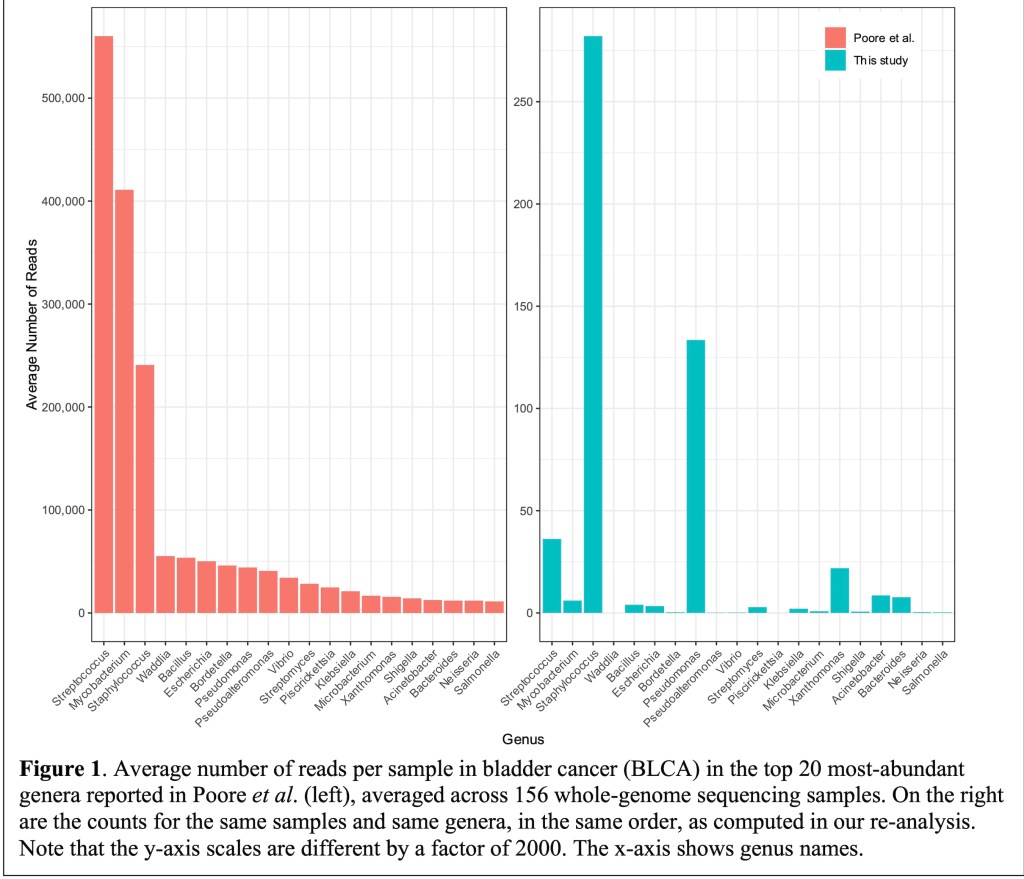
As a result, ca. 1000 times more reads were identified as microbial than the actual numbers obtained by Johns Hopkins researchers with updated references (as seen on Y-scales in their Fig. 1).
Johns Hopkins researchers also traced defects in classifier methodology, notably, converting zero raw counts into biased positive normalized values. I am not sure about the formulae that lead to it, but it can be achieved with quantile normalization applied to lowest ranks when the library size is related to phenotypes. While quantile normalization will remain a standard part of classifying pipelines, for the lowest and highest ranks it produces noise values that probably should be converted to “min-val” and “max-val” in a way that depends on context.
As quantile normalization and its equivalents is such a standard tool, all data scientists should be alerted to its limitations, and the same is true for other standard tools and references.
Nature paper:
Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, Kosciolek T, Janssen S, Metcalf J, Song SJ, Kanbar J, Miller-Montgomery S, Heaton R, Mckay R, Patel SP, Swafford AD, Knight R. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature. 2020 Mar;579(7800):567-574. doi: 10.1038/s41586-020-2095-1. Epub 2020 Mar 11. PMID: 32214244; PMCID: PMC7500457.
Reanalysis:
Abraham Gihawi, Yuchen Ge, Jennifer Lu, Daniela Puiu, Amanda Xu, Colin S. Cooper, Daniel S. Brewer, Mihaela Pertea, and Steven L. Salzberg. Major data analysis errors invalidate cancer microbiome findings. https://www.biorxiv.org/content/10.1101/2023.07.28.550993v1.full.pdf
Alerts about microbial references:
Breitwieser FP, Pertea M, Zimin AV, Salzberg SL. 2019. Human contamination in bacterial genomes has created thousands of spurious proteins. Genome Res 29:954-960.
Steinegger M, Salzberg SL. 2020. Terminating contamination: large-scale search identifies more than 2,000,000 contaminated entries in GenBank. Genome Biol 21:115
